Electrophoresis is a method used to separate macromolecules on the basis of their net (surface) charge and their molecular mass. The protein mixture, dissolved in an appropriate buffer, is applied to a gel or paper strip or other support and an electrical current is applied to promote the migration of the charged molecules towards the positive or negative electrode. It is important to remember that nucleic acids are always negatively charged, and thus migrate towards the positive electrode, whereas proteins may be positively or negatively charged depending on pH. The reason why proteins are electrically charged is that the side chains of some aminoacid residues may have titratable chemical groups: e.g. glutamic and aspartic acids may be present in the neutral (R-COOH) or negatively charged state (R-COO-), whereas histidines or lysines may be present in the neutral or positively charged state. As a general rule at alkaline pH most proteins are negatively charged (i.e. deprotonated) and migrate towards the positive electrode, whereas the opposite occurs at acidic pH.
This short photographic tutorial illustrates the preparation and run of a polyacrylamide gel electrophoresis for the separation of proteins.
Thanks are due to Dr. Adele Di Matteo for the skillful assistance.
The acrylamide solution, mixed with the appropriate reagents and catalysts (sodium persulfate and tetramethylene diamine) is poured between two glass plates, to realize the gel slab.


The protein samples, dissolved in the same buffer as the one used to cast the gel, are made denser by addition of sucrose or glycerol, and a tracking dye (bromophenol blue) is added. They are then deposed in the pits.
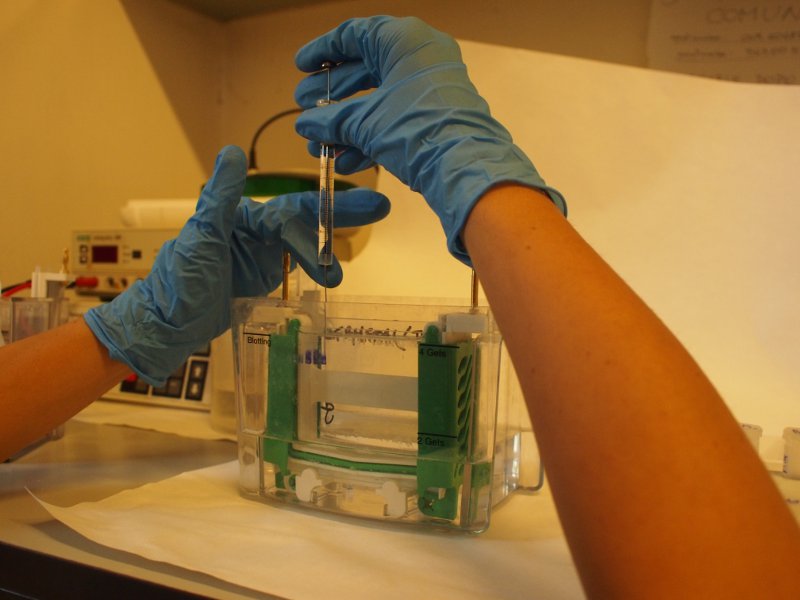

The gel is now loaded and is in contact with the upper and lower buffer reservoirs, which are connected to the electrodes. A current is applied, usually at 3-5 mA/sample and the run is started. The tracking dye, due to its low molecular weight migrates faster than the macromolecules and indicates the moving edge. When the tracking dye reaches the bottom of the gel the electrodes are disconnected and the run is stopped.

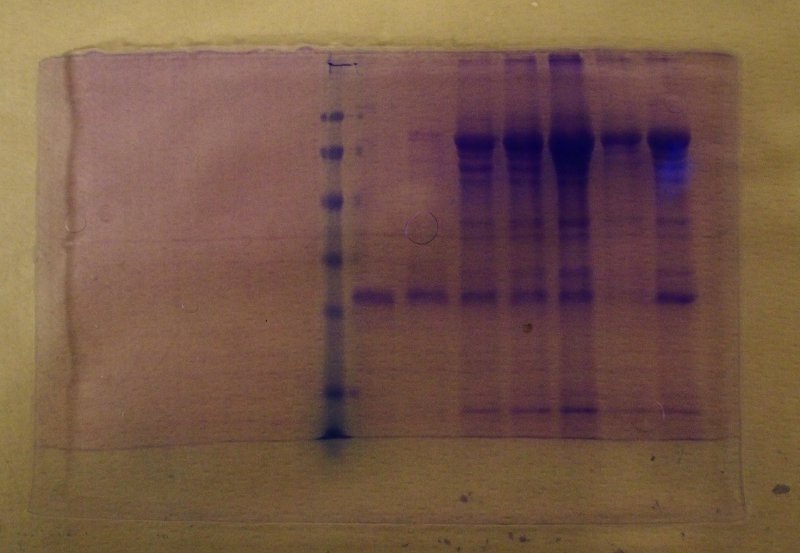
At the end of the run the gel is removed from the glass plates, fixed in a solution of trichloroacetic acid to precipitate the proteins and stained using an appropriate dye (most often coomassie blue, that specifically binds to proteins). After destaining in ethanol and acetic acid, to remove the unbound dye, the proteins appear as blue bands: